In this file (default is
option.txt
), the algorithmic parameters and any other controls are given as:
<parameter> <value> [further values]
These can also be accesses by calling the programm with the
-h
or--help
option. Each parameter is given on a new line. Here is a list of all available parameter options:| Input & Output | |
|---|---|
| output_file | output file (additional to stdout) |
| input_dir | input directory (working directory before run) |
| output_dir | result directory (working directory while and after run) |
| objective | objective function file (default: objectivefunction.txt) |
| bblocks | building blocks file (default: buildingblocks.txt) |
| first_model | initial ”seed” model file |
| save_every | number of generations after which to save models |
| num_save | number of models to save from population every save_every turn |
| Experiment Settings | |
| num_pop | number of populations (default 1) |
| iso_time | isoluation time when using multiple populations |
| mu | population size (default: 10) |
| lambda | number of offspring (default: 100) |
| comma_selection | if given, comma selection is used instead of plus selection |
| mutation_probs | relative frequencies for the different kinds of mutations (default: 1 1 1 1 1 1 1; no mutation, add/delete species, replace reaction, add/delete reaction, duplicate species) |
| max_mutations | maximal number of mutations per mutation-turn (default: 1) |
| cross_prob | probability of crossover instead of mutation |
| pf_settings | population size, number of offspring, number of generations of local parameter fitter (CMA-ES-algorithm, (mu,lambda) strategy) |
| Termination Conditions | |
| stop_time | time limit (in seconds) |
| stop_turn | iteration limit |
| stop_fitness | fitness to reach (default: 0.0) |

The modular architecture allows the user to specify his or her own fitness function in a separate file in the directory
src/objectives
. This file then has to be included into the classObjectiveFunction
(just follow the wayio-table.cpp
is included now). However, a default objective function based on an input-output table is available which can readily be used. A text fileobjectivefunction.txt
has to be provided which specifies the desired input-output behaviour of selected species in the evolved models. In the default case, fitness is then calculated as the mean square difference between the desired and the realised timecourses. An example objective function file is given in figure 4.
The evolutionary software system deals with SBML models as solution candidates for the optimisation problem. Since these can have nearly arbitrary structure, it is necessary to lay down a-priori model specifications that the optimisation process can use. This is done in the file
buildingblocks.txt
and handled in the classBuildingBlocks
. The building-blocks specification consists of the following elements:- The maximum number of species that can be used in the model
- The maximum number of reactions
- Lower and upper bounds for the initial concentration of each species
- A list of allowed reaction mechanisms, given in the following format (on one line):
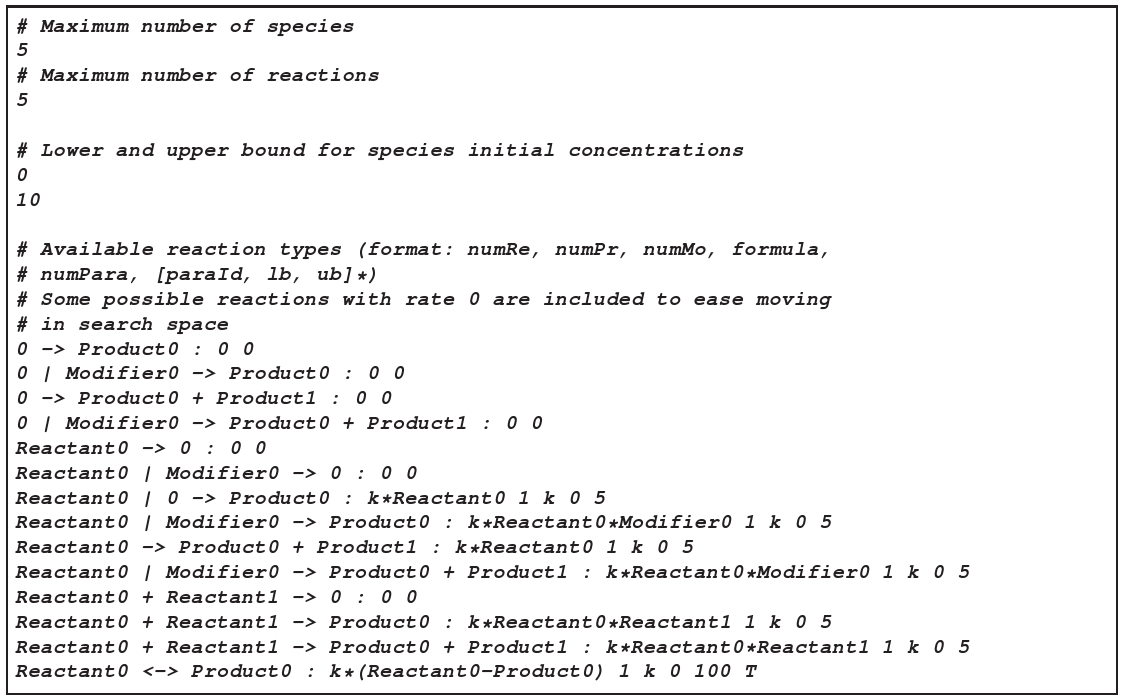
Re1 + Re2 + ... | Mod1 + Mod2 + ... -> Pr1 + Pr2 + ... : kinetic_law number_of_parameters (name lb ub)* [T]where (name lb ub)* denotes a series of parameter names followed by their lower and upper bounds, and the optional flagT
at the end denotes transport reactions, which can involve species from different compartments (others cannot).The reserved namesReactant<num>
,Modifier<num>
, andProduct<num>
can be replaced by arbitrary species in the evolver. This way, a mix of species-specific and general reaction mechanisms can be specified. E.g., the reactionReactant0 + 02 -> Product0describes the oxidization of an arbitrary species, i.e. the reaction of an arbitrary species with Oxygen to another arbitrary species. An example of abuildingblocks.txt
file is given in figure 3.
Importing building blocks automatically
The classBuildingBlocks
provides the methodlearnReactionsFromModel
, which loads reaction mechanisms from a given SBML model into the building-blocks (abstracting from the specific participants in the original reaction). This can be used to construct a library of reaction mechanisms from published SBML models.
For the initial population, the program will load any SBML models in files of type
<modelname>.model.xml
from the current directory, and fill it up with random SBML models containing the number of species and reactions specified in the options, as well as any species required for the objective function. Also, it is possible to specify one initial starting model that is mutated once to create initial solutions (using thefirst_model
option inoptions.txt
). If no initial population is given, random models are created, for which the minimal and maximal size is specified in the options.
To start the evolver on a single machine, run
sbmlevolver [<arguments>]
from the command line. By default, arguments are read from file
options.txt
. If a different options file is wanted, this has to be given directly after the call. After this, any options can be given, overriding the options specified in the optionsfile.
During the run, information about the fitness-development of the population is displayed on the screen. There are two ways to finish the program: either the pre-specified fitness, number of iterations or runtime is reached, or the user sends the signal SIGUSR1 to the process (
kill -SIGUSR1 <pid>
). After finishing, the program stores the population of the last generation in separate SBML files of the formatresult_<id>_<fitness>.model.xml.
With these files, a run can be restarted from the exact point where it terminated.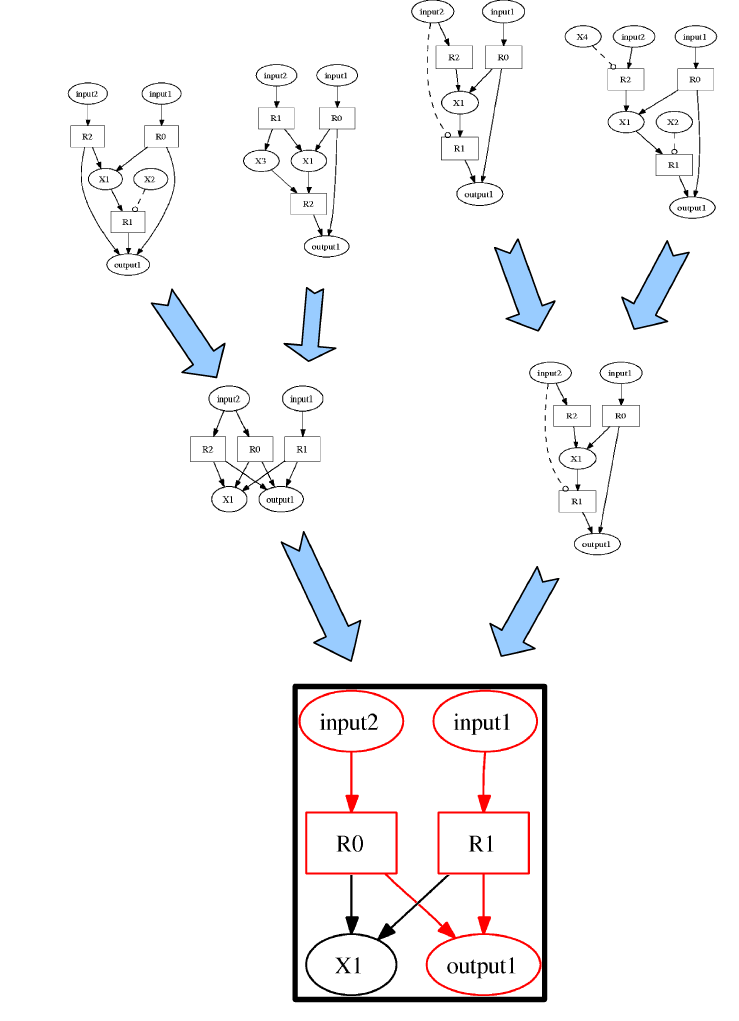
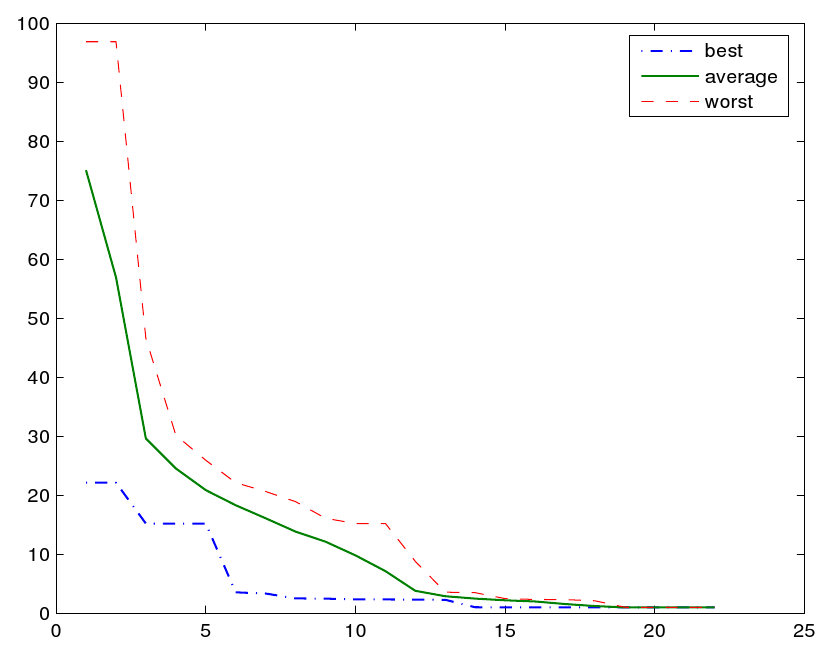
Results of the evolution run are given in two ways: First, running statistics about the population are displayed, such as the best and average fitnesses, and mean numbers of species and reactions. Using the
output_file
option, this can be stored into a file and used for later evaluation. Second, the final population is stored as SBML models, which can be investigated using standard Systems Biology software tools (see list on www.sbml.org). Since the SBML_odeSolver has to be installed anyway, this program (and its associated tools such as the ParameterScanner) can be used to simulate, visualise and analyse the models.
Figure 1 shows a series of snapshots from the evolution of a network capable of adding two numbers, while the corresponding fitness development is shown in figure 2. If the evolution gets stuck in a (supposedly local) optimum, one can stop and restart it with a different set of parameters, using the final population of the first run as an initial population for the second.